By Tracy TreDenick, Head of Regulatory and Quality, and Senior Consultant
Monoclonal antibodies (mAbs) elegant specificity has catapulted them to a starring role within the world of precision medicine over the course of the last couple of decades. As is typically the case with new and revolutionary technologies, there have been fits and starts. The first FDA-approved therapeutic mAb, Orthoclone OKT3, was approved in 1986 to limit organ transplant rejection. Since 1986, more than 70 mAbs, serving either therapeutic or diagnostic purposes, have been approved. There have been many strides forward, as well as setbacks, but mAbs have grown from a small part of the drug industry to now being more than 50 percent of the overall biotherapeutic market. There’s a huge pipeline of antibodies still to be commercialized that will continue to deliver substantial market growth for years to come.
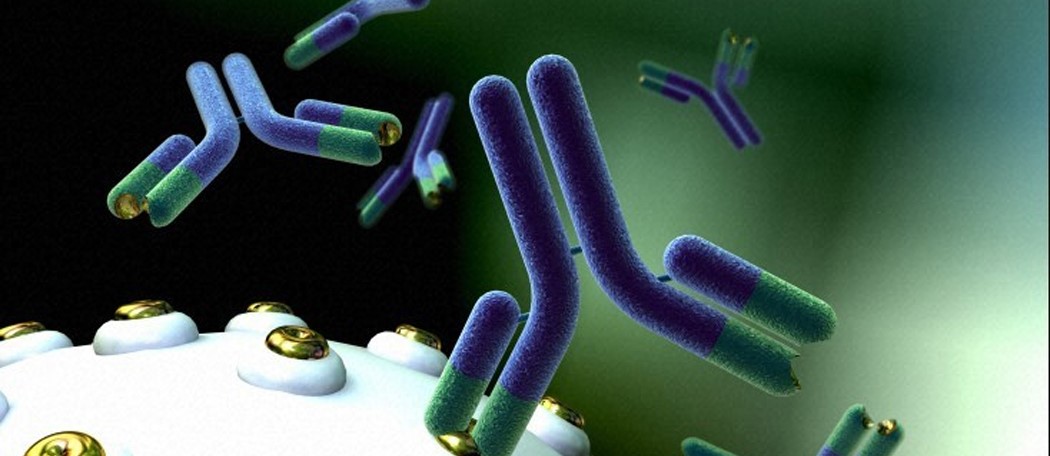
Before diving into the discussion of the benefits, challenges and exciting future of mAbs, it’s worth taking a moment to reflect on merely one aspect of the near magical intricacy of antibodies. Of course, an antibody is a blood protein produced to counteract a specific antigen, or foreign substance such as bacteria, viruses or another foreign substance detected by the body.
mAbs bind monospecifically to one antigen, one epitope, or one cell type making them incredibly useful for highly targeted therapeutic administration. Their therapeutic specificity makes them ideally suited for helping to minimize adverse side effects, especially when highly toxic drug substances must be delivered.
While the concept of mAb therapeutics is a simple one, the application of these concepts is far from easy. This paper explores some of the exciting directions mAb therapy is heading, and the challenges and enabling technologies impacting this therapeutic area.
Monoclonal Antibody Development Challenges and Progress
The development of a successful therapeutic mAb requires the identification and creation of a selective and potent molecule that performs the needed task, humanization of sequences, affinity maturation, Fc engineering to modulate effector functions, and…download white paper