From post-aging performance testing to container closure integrity, robust design controls extend far beyond “constituent part” requirements.
With the increase in complexity of some combination products and co-packaged kits, the need remains for ensuring that the patient gets the right drug at the right dose at the right time. While it is acceptable to make improvements to products as more information becomes available, it’s important to recognize that a change to a co-packaged kit can result in a warning letter if design controls are not properly implemented.
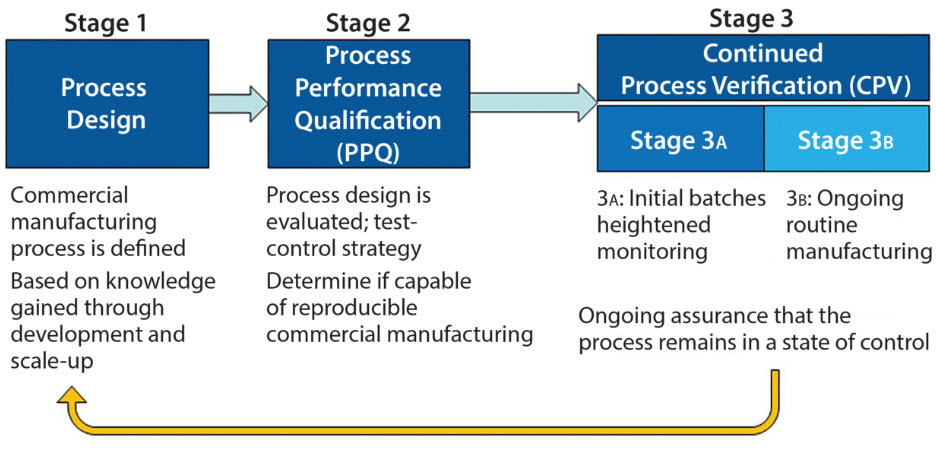
In her presentation at the 2017 PDA Annual Meeting in Anaheim, CA, Tracy TreDenick, Head of Regulatory and Quality Assurance and Founding Partner at BioTechLogic, explained that design controls for co-packaged kits include requirements for individual constituent parts, but also include inter-component dimensional and functional specifications, system integration verification testing and shipping of aged components. “FDA is asking for dimensional and functional specs for the final finished form,” she said.
This is not just about the closure of a prefilled syringe, but about the biocompatibility of all parts combined. She noted, “The challenge is understanding the constituent parts and the system integration requirements.” Continue Reading Article